如何科學、客觀地制訂溶出度試驗質量標準
謝沐風
上海市食品藥品檢驗所 上海市浦東新區張衡路1500號
郵編:201203 郵箱:xiemufeng@sina.com
摘要: 本文詳盡闡述了如何科學、客觀地擬定質量標準中溶出度試驗各參數,并廓清了擬定出發點和控制要素,為如何使該試驗法的擬定體現出制劑內在品質提供了佐證與參考。
關鍵詞: 溶出度試驗法;擬定;參數
溶出度試驗作為“評價固體制劑內在品質的靈魂與核心”,隨著人們對該項技術的不斷研究與深入,對其認識與理解亦在不斷發展與變化著
[,,]。現今,該試驗不僅具有為建立體內外相關性而設立的宗旨,且還已成為證明藥物體內釋放特性的一種簡單、廉價而不失嚴謹的實驗室檢測方法。
尤其“在多種pH值溶出介質中溶出曲線(本文的“溶出度曲線”包涵釋放曲線,以下同)的測定(以及比對)”更是成為“剖析”和“肢解”固體制劑內在品質一種擘肌分理、抽絲剝繭的重要手段;成為固體制劑內在品質呈現于外在的一種“表象”、“映射”與“載體”;成為在仿制藥“殊途同歸”的研發進程中、提高生物等效性(BE)試驗成功率所歷經的“必由之路”,甚**成為該品種在其工藝與處方完全成熟、持續進行多批大規模生產后,所擁有的特定特征“指紋圖譜”。總之,“多條溶出曲線的測定”可在與固體制劑品質相關的所有環節上均發揮出舉足輕重的作用。
無論是研發仿制藥/創新藥,或是省級藥檢所在審核質量標準、起草藥典、標準提高/轉正、評價性抽驗、不同來源的藥品間品質評價等各項工作,均涉及到在進行了大量深入的研究驗證后,**終應如何科學、客觀地制訂質量標準中溶出度試驗法,即通過該法測得的結果能否充分反映出該產品所應具有的內在優良品質、所應具有的良好體內生物利用度以及品質均一性,能否通過該試驗法的制訂促使企業進行深入的制劑工藝/處方研究與周而復始的品質保證等等
[,]。
截止目前,關于如何制訂質量標準中溶出度試驗法的文章尚未有發表。本人已撰寫發表多篇了有關溶出度試驗在藥物研發、質控、內在質量評價等方面如何發揮作用的文章
[,,],本篇將就本人工作經驗與感悟,詳盡闡述如何制訂溶出度試驗質量標準。
一、
針對仿制藥
無論是《藥品注冊管理辦法》中的三類新藥(已在G外上市銷售但尚未在G內上市銷售的藥品)還是六類新藥(已有G家藥品標準的原料藥或者制劑),兩者**區別就是有無“可參照的質量標準”,但均請按照以下思路進行。
1.
既有質量標準的可參照性
針對六類仿制藥,實驗者肯定會查詢到相關參考文獻,如各G藥典、G家藥監局標準、進口質量標準、《日本橙皮書》、美GFDA公布的溶出曲線數據庫、日本仿制藥技術申報資料概要(采用“醫薬品インタビューフォーム”在Google上收索即可,內容僅為日文)等,但由于這些標準本身所具有的局限性、歷史性和利益性,因此jue不建議實驗者不經對原研品的剖析(詳見2~7內容),就直接擷取,即便是G外藥典或進口質量標準,制訂者也不應盲目迷信(詳見討論1)。這正是G家藥品審評中心自2008年起就反復提出“仿產品不是仿標準”宗旨的核心要素之一(對于口服固體制劑,還有一要素為“有關物質”)。
2.
購買原研品
六類仿制藥,均可購買到進口品或原研廠在G內合資企業產品。
三類新藥(其實也是仿制藥),研發者必須想盡辦法購買來原研品,可通過私人關系帶入,或通過“世界藥房”網站、“白求恩藥房”網站等中介,還可向G家藥監局注冊司申請“一次性進口”批件(少量、僅用于研發)等多種方式。jue不應以購買不來為借口,“天馬行空、閉門造車”地進行處方開發與制劑工藝研究;且購買時應盡可能獲取不同時間段的多個批號樣品(如出廠不久的和臨近效期的)。
將購買來的原研品作為0個月計,放入冰箱內冷藏。對于研發者,強烈建議與仿制品同時進行6個月穩定性考核,每一時間點樣品取出后均放入冰箱冷藏,直**6個月結束。shou先測定**終時間點的多條溶出曲線(當然還有有關物質),觀測與0個月相比的變化情形,再酌情考慮是否需測定其他時間點樣品,從而根據原研品溶出曲線波動情況對自身仿制品的研發和品質做出正確判斷與準確評估。
3.
采用多條溶出曲線“循序漸進、抽絲剝繭”般剖析原研品
在進行溶出曲線測定前,一定要先進行以下三項工作
[4,]。
3.1
“pH值-溶解度”曲線的測定
取過量原料藥(可為預經微粉化處理),置8支具塞試管中,分別加pH1.0、…、8.0溶出介質適量,置37℃水浴振蕩過夜,形成過飽和溶液,取出后濾過,取續濾液經HPLC法測得溶解度,繪制曲線。對于難溶性藥物,對照品溶液可酌情采用純甲醇或純乙腈配制。
該曲線的繪制可提供眾多信息:如與X軸平行,說明各pH值溶解度幾近一致,由此可預測多條溶出曲線應重合;如曲線上有陡峭變化、甚**是有數量級差異,則可揭示多條溶出曲線形狀必會有所差異(即高低錯落),**低的那條曲線一定是溶解度**低的pH值介質,這為制劑研發方向提供了針對性與佐證素材。
當出現主成分在某pH值介質中極不穩定、溶解后即迅速分解,無法測定的情況,則該介質溶解度可不測定,其溶出曲線亦可免做。
3.2
pKa值的查詢與測定
pKa值的知曉也十分重要,可通過查詢或測定獲得。測定法可參照以下三篇文獻【,,】。若該值未涵蓋于四條標準溶出曲線pH值范圍,則研發時第5~6條溶出曲線就應測定該pKa值所對映的pH值介質或以上“pH值-溶解度曲線”上急劇變化的pH值,這些pH值溶出曲線的測定對于仿制制劑研發和曲線比對均具有十分重要的意義。
3.3
主成分在各溶出介質中的穩定性
為獲得準確試驗數據,該驗證必不可少。
建議取原料藥粉末配制的溶液即可,無需取樣品溶出液。
3.4
采用溶出曲線“剖析”與“肢解”原研品
本人已發表此篇文章,請詳見參考文獻[]。
4.
質量標準中各參數的制訂[4,]
在進行了以上對原研品和仿制品多pH值溶出介質中溶出曲線的測定后,才能科學客觀、合理全面地制訂質量標準。具體闡述如下:
4.1
溶出介質的選擇
4.1.1
速釋制劑
shou先應滿足在該介質中**終溶出量達85%以上,然后可按下列情況分別選取
[]。
Ø 根據該藥物在體內吸收主要消化道部位的生理pH值(適用于一般情況);
Ø 能在一定程度上反映體內外相關性的pH值(適用于創新藥);
Ø **能體現不同來源制劑間彼此差異的pH值(適用于標準轉正/藥典起草、尤適合于我G大量仿制藥存在情形);
Ø **能反映生產工藝變化、偏差的pH值(適用于企業內控標準,用于批間樣品品質均一性的評價);
Ø 溶出曲線中**低的pH值(適用于企業內控標準,用于應對G家市場檢查與監督);
Ø 溶出數據精密度更佳的pH值(某些樣品在**介質中精密度不佳時、更換為精密度更為理想的介質)
當多條溶出曲線重合時(各時間點溶出量相差不超過10.0%),《日本橙皮書》傾向**“水”。其出發點為:雖然水的pH值和表面張力會因來源不同而改變,且在試驗過程中也可能會因藥物影響或者溶解入二氧化碳使溶出行為發生變化,但考慮到發生上述可能性的概率較小,且可通過事先驗證予以探明,故秉承環保、提高試驗效率等出發點,質量標準中**水。而美G藥典鑒于以上問題的存在,傾向采用帶有pH值的介質。筆者更傾向日本作法,因水的pH值范圍5.0~7.0已被上述多條溶出曲線的pH值所涵蓋。
當多條溶出曲線不重合,則可根據上述各針對性酌情擬定。
4.1.2
腸溶制劑
應規定酸中介質(pH1.0~1.2)和堿中介質(pH6.8~7.2)釋放量的測定。
酸中釋放量的測定現今愈發傾向通過測得準確數據予以表達、而非肉眼觀察外觀形狀進行控制(日本橙皮書皆采用此法),通常規定2小時不得過10%。為防止藥物在年輕人體內發生過量釋放,甚**在該階段可故意采用高轉速(如100轉),以進一步探明藥物的內在優良品質。
如主成分溶出后在酸性介質中不穩定旋即降解,即便立即測定也無法準確評估時,建議測定溶出杯中剩余固體顆粒所含主成分量,隨后用測得百分含量減去剩余百分含量,再除以測得百分含量,即為酸中釋放百分量。
現今,隨著市場上銷售的腸溶衣輔料已皆可在pH1.0~3.0范圍內包裹住藥片,使主成分釋放量合格,而經研究表明:人體內胃環境,隨年齡增長胃酸分泌逐漸減少(胃內pH值范圍1.2~7.6),如此再在pH1.0~1.2范圍內測定已顯得毫無意義,故現今開始逐步測定pH4.5介質,規定依然是不得過10%溶出量。英G藥典自2008年始
[],“奧美拉唑腸溶膠囊/片”的制訂原則便是以此為依據,這樣的測定更有針對性和實用性,值得我們學習和借鑒!
堿中釋放量同速釋制劑;但需注意的是:腸溶衣對紫外測定時常有干擾,故強烈建議采用紫外-兩點相減法或HPLC法,否則極易出現溶出量均值高于含量3.0%以上的情況。
4.1.3
緩控釋制劑
shou先應滿足在該介質中**終溶出量達80%以上。
當體外多條釋放曲線重合時(酸中僅測定2小時即可),建議**水(pH5.0~7.0)作介質,既經濟又方便。jue不建議采用酸性介質,因任何人體內的十二指腸**小腸消化道器官是不存在該值的;也不建議參照人體內消化器官的標準值(pH1.0~1.2、4.0~4.5、6.8)采用不同時段、不同溶出介質(不斷調試)的費時費力方式、且此方式實驗誤差較大。
當體外多條釋放曲線不重合時,建議選擇**終溶出量達80%以上的、**慢的那個pH值介質。
4.1.4
介質中胃蛋白酶和胰蛋白酶的加入
一般情況下,不建議溶出介質中添加這些酶
[5]。但如某些藥物必須飯后服用、且生物等效性試驗需在“進食”狀態下進行時,則在仿制藥研發中必須進行“溶出介質中分別添加胃蛋白酶和胰蛋白酶的比對研究”,此時質量標準中亦應加入。
另:當硬膠囊劑使用囊殼為明膠膠囊時,為避免產生交聯現象影響溶出,可也加入,但需進行驗證。
4.2
取樣時間點與限度的制訂
4.2.1
普通制劑
以**次出現溶出量均在85%以上兩時間點,且該兩點溶出量差在5%以內時(即出現“平臺期”),取前一時間點作為質量標準中取樣時間點,并將該點溶出量減去15%作為溶出限度。這就將“之前我們通常認為的‘溶出度取樣時間點常選擇溶出曲線拐點處后推10~20分鐘’
[]”的原則予以明確化和具體化,因為“拐點處后的10~20分鐘”即為溶出飽和“平臺期”。由此便可推斷出:溶出限度一般僅有70%、75%、80%、85%四個數值。如**時間點為20分鐘,由于其不為刻鐘的整數倍,一般提高**15分鐘,但限度可僅減去10%。
當出現采用50轉、溶出曲線緩慢上升、**60分鐘后才出現平臺期(研究時需測定到360分鐘)時,會出現取樣時間點過于滯后的情形,這不利于產品的日常檢驗。此時為提高試驗效率,可適當放寬參數(如加大轉速)或采用溶出更快的介質,使60分鐘前出現平臺期,即不建議擬定60分鐘后的取樣時間點
[4]。但前提是,這些調整“不應弱化針對不同來源制劑間內在品質的區分力(如采用增加轉速方式)”或“無法建立體內外相關性介質(更換的介質也必須具有體內外相關性的特質)”。
4.2.2
緩/控釋制劑
應**少設定3個時間點(服用方式**2次)或4個時間點(服用方式**1次):**點為避免“突釋”,應設定為試驗1~2小時后或溶出量相當于標示量10~30%時間點;第二點是為考察藥品溶出特性,該限度應設定在溶出量約50%時間點;**后一點是為確保藥物溶出量超過80%時間點。如擬定4點,第二、三、四時間點的溶出量應分別約為40%、60%和80%溶出量。
任何一點的擬定范圍均應勿超過標示含量的20%,且各點溶出限度交叉范圍建議勿超過5%,除非體內特征可顯示出相應的可接受性和波動性。對于零級釋放產品,因其釋放曲線為“一條直線”,故還應增加每小時釋放量的規定(即斜率規定),“硝苯地平控釋片進口質量標準(拜耳出品)”中就有6~8%/小時的嚴格規定。
4.2.3
治療窗狹窄藥物
為防止“突釋”,愈發傾向采用兩點法測定。一可采用“5或10分鐘的**點溶出量不得大于某一限度(以控制突釋),第二點規定一定時間內溶出不得小于某一限度以保證溶出完全”的作法(如《日本橙皮書》中卡馬西平片擬定為5分鐘不得過60%和30分鐘不得少于70%);二可采用緩控釋制劑擬定法:**點規定5或10分鐘時溶出量為一限度范圍而非一上限,以保證其溶出曲線呈現“緩慢上升性”(如美G藥典卡馬西平片規定:10分鐘釋放量應為30~50%,45分鐘時不得少于75%),該規定亦可有效防止處方中加入大量表面活性劑或增溶劑的“投機取巧”作法。
各G制訂的技術指導原則中收載的此類藥物清單如下:氨茶堿、茶堿、膽茶堿、雙羥丙茶堿、苯妥因鈉、丙戊酸、炔雌醇/孕酮制劑、地高辛、洋地黃毒甙、華法令鈉、甲磺酸異他林吸入氣霧劑、卡馬西平、可樂定透皮貼劑、磷酸丙吡胺、硫酸胍乙啶、硫酸奎尼丁、硫酸哌唑嗪、硫酸異丙腎上腺素、米諾地爾、撲米酮、碳酸鋰、鹽酸克林霉素、鹽酸可樂定、鹽酸普魯卡因胺、左甲狀腺素鈉、環孢霉素A、他克莫司、西羅莫司、丙戊酸/丙戊酸鈉等。
4.3
裝置的選擇
建議**通用性強、耐用性好、廣泛普及的籃法與槳法。
膠囊劑**籃法、片劑**槳法
[]。
對于不易采用籃法(如發生堵塞籃法孔隙、或樣品塌陷于藍底、或粘附于籃頂)、且易漂浮于液面的制劑,可考慮采用槳法、加沉降藍;尤對于采用了粘附性較強輔料的緩釋制劑,極易發生試驗過程中始終粘附于溶出杯不同部位、而使溶出數據均一性較差的情況時,更推薦采用沉降藍。jue不建議采用自制沉降裝置,因其制作的不規范性會導致數據的難以重現性。
不推薦使用非法定或非標準試驗裝置。如卻有必要采用,但應提供詳盡驗證資料和充足理由,并充分驗證其必要性與質量可控性(即實驗室間的可操作性、耐受性和易重現性)。如美G藥典第三~七法,決不能因價格昂貴而自行設計組裝,且由于其現階段該儀器尚難以推廣和普及,故筆者建議勿采用。如經典的籃法與槳法無法正確表達出該樣品體外溶出特征,可在此基礎上進行改裝,如針對栓劑或陰道制劑的體外溶出度研究與測定,即如此(詳見參照文獻[,])。
4.4 轉速的擬定
4.4.1 既有觀念的錯誤
由于50轉的攪拌強度被看作與中老年人體胃腸道蠕動強度相當,且由于患者大多為中老人群,故**該轉速。現今G內仍存在一種普遍看法,認為槳板法/50轉≈轉籃法/100轉
[],其實這是錯誤的。筆者在日本G家藥檢所進修期間,經導師指教、并親自采用USP溶出度校正片實驗驗證,結果為
[]:
l **弱級:槳板法/50轉≈轉籃法/50轉(樣品在籃內)≈轉籃法/100轉(樣品在籃外、即沉于杯底)
l 中級:槳板法/75轉≈轉籃法/75轉(樣品在籃內)≈槳板法/50轉(樣品置于沉降籃內)
l **強級:槳板法/100轉≈轉籃法/100轉(樣品在籃內)≈槳板法/75轉(樣品置于沉降籃內)。
猜測可能是當年我G引入溶出度試驗時將英文譯錯所致;截止目前已有不少同仁來電來函反映上式的正確性。當樣品置于沉降藍內,在試驗過程中始終處于杯底,由此受到的外來渦旋力將大于樣品任意漂浮或停滯于杯中某處,故其級別比不置于沉降藍內高出一級。
4.4.2 需放寬試驗參數時
不能隨意采用高于50轉速,因為這將極大地弱化對不同制劑/處方溶出行為的區分力。對于有必要放寬溶出度試驗參數的情況,應分別考察轉速為75~100轉和加入表面活性劑兩種方式,以更好地建立起體內外相關性和能區分不用來源制劑內在品質優劣。
目前傾向于采用“不提高轉速、加入表面活性劑”方式,因對于中老年患者,雖胃腸道蠕動較弱,但體內存在膽堿類物質,可用表面活性劑表達。但當樣品在杯底出現錐度堆積、呈“小山狀”時,則傾向加大轉速、而非加入表面活性劑方式。
表面活性劑添加濃度研究應從0.01%(w/v)起板、按照1、2、3、5級別逐步增加去“剖析”原研制劑(即逐步試驗0.01%、0.02%、0.05%、0.1%、0.2%、0.5%、1.0%、2.0%和3.0%,9個濃度;另:配制溶出介質、溶解表面活性劑以及無機鹽時,一定要采用加熱煮沸方式,切忌采用振搖或超聲),不建議采用3.0%以上濃度。當兩級別間溶出曲線差異較為顯著時,則應適當增加中間濃度以進一步研究。需強調的是:jue不建議不經以上“循序漸進”,而根據“漏槽條件”推斷出某濃度便直接擷取(詳見討論5)。
研究時應注意不同來源的表面活性劑可能會對試驗結果帶來顯著性差異情況,如十二烷基硫酸鈉(SDS)的使用,現今愈發傾向在研究時采用市場某一主流品牌(分析級),并在質量標準中注明,以使其后試驗的重現,jue不建議研究時采用市場上多個品牌、在質量標準中不注明的作法,費時費力、且導致試驗結果無法預料。
綜上所述,若質量標準中未制訂50轉或采用了添加表面活性劑的介質,就應在研發資料和起草說明中予以詳盡驗證和闡述。
4.4.3 **極端試驗條件
質量標準中擬定的**極端條件建議為:槳板法/100轉、溶出介質中加入3.0%表面活性劑。因目前上市的所有原研制劑,皆未有在此條件下沒有一個溶出介質達不到85%(或80%)溶出量的產品;且在該條件下,如任何一個介質中皆未有85%以上溶出量,亦可推斷該制劑猶如“石頭般堅硬”,如此制劑在人體內的生物利用度也就可想而知了,這一點對于創新藥的制劑研發具有很強的指導意義。
溶出介質中嚴禁添加有機溶劑。因為這將極大地降低體內外相關性,嚴重背離溶出度試驗應用理念
[5],是典型的“為了溶出合格而設定”的標準;同時對于人體而言,是不可能“先喝2兩白酒再吃藥的”。遺憾的是,G內的一些質量標準仍有這樣的品種存在,值得深思。
若確實找不到一個理想溶出條件,說明現今的制劑手段尚無法解決該原料藥的溶解性,尚無法開發成適合人體吸收的制劑,此時則應果斷、明智地放棄,使之休眠,直**開發出可解決其生物利用度的輔料。
4.4.4 特例
pH值特例:鑒于3.0%表面活性劑的加入,對于配制和試驗操作皆會帶來極大困難,此時若在pH大于8.0的介質中(即溶解度顯著提高的pH值)可找到一個較為溫和試驗條件下**終溶出量達85%以上,則推薦采用該pH值,即便該值已背離人體正常生理值
[5]。美G藥典中“阿維A酸膠囊(Acitretin Capsule)”采用“籃法/100轉,pH9.6~10.0緩沖液中加3.0%SLS(十二烷基磺酸鈉)作溶出介質,900ml,30分鐘,85%限度”的條件就是出于該考慮。
轉速特例:不推薦采用低于50轉的條件,除非針對特定劑型或特定工藝。如中G藥典“布洛芬緩釋膠囊”擬定30rpm。此時則應格外注意儀器適用性,曾發現不同儀器間測定結果存在顯著差異情況(即低轉速下顯示出儀器間性能差異性)。又如懸浮劑一般采用25~50rpm,但對于出現錐度堆積的情況,可通過適當增加槳速予以改善。
4.5 體積選擇
統一設定為900ml或1000ml。900ml與人體消化道內的體液總體積較為接近,1000ml則是為便于計算。其他體積均無必要選擇。
**于中G藥典收載的第三法 —— 小杯法,是特定歷史時期產物(當時液相不普及,對于小規格制劑,即便采用5cm吸收池,仍無法滿足紫外吸收度應達0.2的**低要求,于是在第二法的基礎之上衍生改良而來)。由于該法不滿足當初設計
溶出儀時所追求的漏槽條件,更不符合大杯法所應具有的流體力學特征,故截**目前其他各G藥典均未采用。現今,隨著液相色譜儀在G內已完全普及,即便再小規格,亦可通過各種措施解決準確測定問題,故該法已無必要。因此,即便既有G內質量標準擬定該法,因原研制劑在研發和質量控制時從未采用過該法,故強烈建議實驗者一律改為大杯法。
5.
投樣量
之前曾有為滿足樣品**低定量限需要,采用一杯內投入2片作法,現今采用HPLC法已皆可解決
[],故投樣原則必須遵循單片樣品方式
[21]。
對于多計量的顆粒劑、混懸液、干混懸劑等劑型,可采用一次單位服用量投入。投入方式可采用:混勻后立即傾入已標化的、帶有刻度的試管中5~10ml,即刻用滴管加樣品**刻度,傾入溶出杯中,并用溶出杯中液體清洗試管。
6.
測定法[]
無論采用何法檢測,均應牢固樹立“研發階段測定法與質量標準測定法應區別對待”的科學理性理念。
6.1 研發時測定法
研發時,溶出測定工作量十分巨大,具不完全統計:速釋制劑仿制藥研發需測定約500條溶出曲線、緩控釋制劑需測定約1000條。如采用紫外法測定,就會有因需不斷篩選處方而采用各種/各來源輔料對紫外測定干擾的擔憂和為滿足紫外檢測需達0.2~0.8吸收度要求、而采取繁瑣稀釋步驟等的顧慮,故強烈建議采用HPLC法。關于如何高效準確、快速便捷地測定大批量溶出度樣品請詳閱參考文獻[23],如此便可省時省力、事半功倍。
目前市售的G產“光纖全自動測定
溶出儀”、將光纖探頭插入溶出杯中直接測得一條完整曲線(可每隔20秒測定),雖是采用紫外法測定,但由于其數據處理程序中已設定了各種校正法去確保排除各種情形的輔料干擾,故值得肯定與推薦。
6.2 質量標準測定法
質量標準檢測僅是一個介質、一個時間點、一個限度,工作量很小,故為考慮測定的方便性,在排除輔料干擾的前提下,可考慮采用紫外法測定;但如含量測定為液相,筆者則建議采用液相,尤其是對于膠囊劑、薄膜衣片、腸溶制劑和緩控釋制劑(囊殼、薄膜衣、腸溶衣、緩控釋制劑輔料極易干擾紫外測定)更是如此
[]。
溶出度均值如超出含量2.0%以上,則說明有干擾,此時就會造成不合格樣品誤判為合格情況的出現,必須予以查明和更改測定法。目前,G內出現此種情況的品種愈發增多,蓋因采用了價廉劣質的輔料所致,值得關注和警示。
7.
復方制劑
對于復方制劑,在遵循以上原則基礎上靈活選擇,不必拘泥于采用同一套參數。可根據各組分情況,予以針對性擬定
[]。測定法**液相。
8.
如何正確看待驗證結果與既有質量標準
根據以上2~7內容 —— 如何制訂溶出度試驗質量標準原則,如驗證結果與既有質量標準相符,則可參照采納;如不一致,則應制訂更為科學、合理的溶出度試驗參數與測定法。擬定者決不能墨守成規、畫地為牢,在知曉了既有質量標準不科學性、不客觀性的前提下,還照搬照抄、以訛傳訛!
9.
質量標準的漸進性與完善性
應強調指出的是:任何一個質量標準都不是一成不變、不可更改的!研究者可根據產品在不同階段出現的特性和隨著對該產品研究的不斷深入,對質量標準中溶出度試驗條件予以科學、客觀的更改與完善。甚**在使用溶出度試驗進行產品質量控制時,亦可根據不同情形和需要采用不同方法。
l 如申報生產時,隨著制劑工藝放大和處方優化,更改申報臨床時的溶出度試驗參數;
l 如省級藥檢所擬定藥典或標準轉正時,可針對不同來源樣品、擬定**具區分力的溶出介質;
l 如企業內控標準,為保證批間樣品均一性,在完成既有質量標準規定檢測的溶出介質后,增加其他更能反映批間樣品波動性的溶出介質等等。
10.
溶出度試驗質量標準擬定理念
由于溶出度試驗是口服固體制劑的“靈魂”與“核心”所在,故如何科學、合理的擬定**關重要。其根本出發點為:制訂高標準、嚴要求的溶出度試驗質量標準來撬動制劑的深入研究,尤其是工藝藥劑學的研究與實踐。我G藥典或G家標準中部分品種出現的“加入有機溶劑”、“150轉轉速”、“ 加入高濃度表面活性劑”等,皆是與以上理念背道而馳的,蓋因這些品種低劣的制劑工藝所致,只好通過放寬溶出度試驗條件來滿足,但此種藥物在人體內的生物利用度可想而知了!
二、
針對創新藥[5]
創新藥質量標準中擬定溶出度試驗的主要目的是為避免出現那些生物利用度不佳的處方/工藝制得樣品情況發生。如主成分在各pH值溶出介質中溶解度幾近一致,則體外溶出行為的追求目標一定是各條溶出曲線重合。如主成分在各pH值溶出介質中溶解度相差較大,則體外溶出行為的追求目標應是盡可能地“抬高”低溶解度的那條溶出曲線,并力爭使其與其他曲線重合。
shou先篩選出生產工藝和處方輔料中影響生物特性的若干個關鍵性因素(如4個因素),通過正交設計或均勻設計得到數個處方(如10個),小試制得各處方樣品,分別進行體外溶出曲線測定,根據以上目標推斷出4個關鍵性因素的篩選方向,按此方向,再次通過正交設計或均勻設計得到10個處方構成,再次進行溶出曲線測定,如此重復下去進行30~50個處方研究后,應可尋找到:在某一介質條件下能夠區分出這些制劑溶出的優劣,即辨明“好、中、差”三個處方。然后通過動物試驗(可以從小鼠過渡到大鼠,再過度到Beagle犬)驗證這三種處方在動物體內生物利用度的差異性,并**好與體外溶出曲線的差異性建議起相關性(兩者可不斷趨同);**后規模化生產出**為優良處方的制劑進行新藥臨床試驗驗證后,按照以上思路制訂該產品質量標準中的溶出度試驗參數即可。
三、討論
1.
非pH值依賴型制劑 —— **制劑的**表達。
經研究表明:隨年齡增長,人體內胃酸分泌逐漸減少,正常人群胃內pH值1.2~7.6、十二指腸pH值3.1~6.7、小腸pH值5.2~6.0
[],可謂千變萬化。一個優良制劑,應在人體內無論何種pH值環境下均有較為相同的釋放行為,從而保證在各種環境下皆有吸收、皆有目標生物利用度。尤對于緩控釋制劑,由于在體內停留時間長、“途徑”各種pH值,更應具備此種“特征體外表現”。這便是制劑研發人員所應追求的“**制劑的**表達 ——體外多條溶出/釋放曲線重合”目標,即應盡可能研制出“非pH值依賴型制劑”。雖然這一目標某種程度上受到溶解度或輔料性能影響,但經對眾多原研品的測定表明:jue大部分藥物均具有以上這些溶出特征,可見原研品在制劑研發上的投入與深入,而我G現今已上市的仿制藥在該體外表觀上相差甚遠,其主要差距其實是對工藝藥劑學的認知不足與該方面人才的匱乏。這也為我們指明了努力方向與奮斗目標!
2.
既有質量標準的局限性、歷史性和利益性
2.1G內既有質量標準
由于眾多歷史原因和制訂/審核人員當時認知的局限性,部分品種的質量標準是秉承“溶出度跟著制劑走”—— 即為了讓該品種所有G內生產企業產品均合格;為了找到一個緩控釋條件而體現緩控釋性特性,這樣一個背道而馳的錯誤理念制訂而成。從已發表的眾多溶出度研究文章和自2008年G家藥監局開始實施的“全G評價性抽驗所進行的探索性研究結果”中已充分體現,此處不再贅述。
2.2 英G(BP)/歐洲藥典(EP)
無論是原料藥還是制劑的有關物質檢測,該G藥典均將各雜質、色譜條件、系統適用性等關鍵性參數羅列得清清楚楚、一目了然;但收載的制劑卻很少、口服固體制劑更少,甚**有些難溶性口服固體制劑、緩控釋制劑竟然未制訂溶出度檢測項
[]。蓋因溶出度試驗作為評價口服固體制劑內在品質優良與否的核心指標,不便公開而已,故索性不予登載。
2.3 日本藥典(JP)
表現形式完全同英G/歐洲藥典,即便收載了溶出度試驗項,也往往標注為“略”或“詳見局外規”。蓋因日本藥品審評部門和G家藥檢所意識到溶出度試驗重要性后,伴隨1998年開始實施的“藥品品質再評價工程”,將溶出度試驗質量標準單獨匯編成《日本橙皮書》(亦稱《局外規》)單獨出版。
2.4 美G藥典(USP)
是各G藥典中溶出度試驗**具參考價值的文獻,但我們亦應了解其產生過程與背景:USP一般在該藥品行政保護到期前向原研企業和各仿制企業(FDA已批準、尚未上市銷售)發出邀請函告知將收載該品種,希望各單位提供質量標準,當各單位提供的溶出度試驗參數不同時(仿制藥“殊途同歸”的體現),USP不予統一、全部登載,因為這些標準均經FDA審核通過,產品皆為生物等效,這就是我們有時會看到個別品種會登載數個溶出度試驗條件的原因所在。一般情況下:TSET1為原研品。如僅登載一個條件,一般也是由原研企業提供的。
然而,即便如此亦不建議制訂者直接擷取,因原研品自上市到**保護期結束期間,歷經大量的、各人群的臨床試驗驗證,原研企業可能仍在不斷針對處方與工藝進行優化和完善,且樣品生產規模也早已遠大于**初臨床申報時(質量標準是那時產生),因此有些品種的質量標準已不能準確、客觀地反映現今上市品所具有的內在優良品質,故筆者仍是強烈建議參照前述試驗思路 —— 必須購買來現階段上市品進行研判。如“尼莫地平片”,BP和USP皆擬定溶出介質中添加0.3%表面活性劑,中G藥典完全相同,可經試驗發現:原研品在0.0~0.3%溶出介質中,溶出曲線幾乎沒有任何變化(**終溶出量皆達85%以上,完全可以不添加),而G內眾多仿制品卻迥然不同:為0.0%溶出介質時,釋放量很少。這充分啟示我們:對原研品的質量評估不能畫地為牢似地照搬照抄既有質量標準。
2.5 《日本橙皮書》[]
日本自1998年開始實施“藥品品質再評價工程”以來,在《仿制藥生物等效性試驗指導原則》、《規格不同的口服固體制劑生物等效性試驗指導原則》、《口服固體制劑處方變更后生物等效性試驗指導原則》和《固體制劑改變劑型后生物等效性試驗指導原則》等一系列指導原則出臺的背景基礎上,開始陸續出版《日本醫療用醫薬品品質情報集(即日本橙皮書與參比制劑目錄,官方網址為:http://www.jp-orangebook.gr.jp/)》。截止2010年底,共進行了670個品種的編撰工作,其中詳細羅列了每一品種的“解離常數”、“在4種pH值溶出介質中的溶解度”、“在各pH值溶出介質中的溶液穩定性”(“3. 采用多條溶出曲線循序漸進剖析原研制劑”項下已述)、“4條標準溶出曲線”、“溶出度試驗質量標準”,一些品種還有“主成分對照品質量標準”、“雜質對照品質量標準”等信息。2011年1月,G家藥品審評中心組織翻譯了這些品種,并在其官方網站(www.cde.org.cn)主頁右側建立了《日本溶出曲線數據庫專欄查詢系統(中文翻譯版)》
[]。
2.6美GFDA公布的“溶出曲線數據庫”
美GFDA-CDER(藥品審評中心)屬下的仿制藥辦公室里的生物等效部(by the Division of Bioequivalence, Office of Generic Drugs),為提高仿制藥內在品質、強化各項審評規范,從2004年初開始效仿日本,從多種溶出介質中遴選出**能反映內在品質的一個溶出介質匯編成集,登載于該部官方網站:http://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfm,并于每季度更新一次。截止2011年**季度,已進行了約900個品種的披露。列舉如下:
|
Drug Name |
Dosage Form |
USP Apparatus |
Speed
(RPMs) |
Medium |
Volume
(mL) |
Recommended Sampling
Times (minutes) |
Date Updated |
|
Abacavir Sulfate |
Tablet |
II(Paddle) |
75 |
0.1N HCl |
900 |
5, 10, 15, and 30 |
03/22/2006 |
同時,該部歡迎大家來電來函,對收載的參數提出異議和修改意見。關于該數據庫的說明與答疑內容亦十分重要(http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm073197.htm),請讀者自行瀏覽,此處不再贅述。
2.7 進口質量標準
由于溶出度試驗的重要性,不排除會有一些企業故意放寬溶出度試驗參數,從而確保進口的各批樣品均符合規定的“利益性色彩存在”。這實際上也對我們的審評員和試驗復核人員的技術水準和專業把握提出了更高要求。建議仍是秉承以上思路進行研判。
3.
質量標準中可不擬定溶出度試驗品種和仿制藥研發時可申請豁免生物等效性試驗品種
口服固體制劑質量標準中并非一定要擬定溶出度檢查項,當原料藥屬于寬治療指數藥物、并為生物藥劑學分類系統(BCS)**類藥物——高溶解性、高滲透性,且采用槳板法/50轉,制劑可在**少四種溶出介質中均達30分鐘85%以上溶出量,同時輔料量與主藥量相比未超過30%,輔料中也未加入表面活性劑、甘露醇和山梨醇
[](因為這些輔料可造成溶出度試驗對生物等效預測的誤判)時,質量標準中就可不擬定溶出度檢查,僅采用崩解時限予以控制
[]。因為此類制劑在胃內(任何pH值)的崩解、吸收已不受胃排空時間的影響。拉米夫定片(0.1g規格)
[]質量標準便如此。
當原研品屬于此類制劑時,仿制藥研發亦必須具有以上各特性,此時則可申請豁免生物等效性試驗,不過建議溶出曲線的測定應**少為5~6條
[24]。世界衛生組織于2010年11月公布了31個此類藥物清單,并對每一藥物予以了詳解
[]:對乙酰氨基酚(撲熱息痛)、乙酰唑胺、阿昔洛韋、鹽酸阿米替林、阿替洛爾、磷酸氯喹、硫酸氯喹、鹽酸氯喹、西咪替丁、鹽酸環丙沙星、雙氯芬酸鉀、雙氯芬酸鈉、鹽酸強力霉素、二鹽酸乙胺丁醇、呋塞米、布洛芬、異煙肼、拉米夫定、左氧氟沙星、鹽酸甲氟喹、甲硝唑、鹽酸甲氧氯普胺、強的松龍、強的松、鹽酸普奈洛爾、吡嗪酰胺、硫酸奎納定、鹽酸雷尼替丁、利福平、鹽酸維拉帕米、甲硝唑。
藥物滲透性可通過某些網站查詢,如美G口服藥物傳遞研究公司(Therapeutic Systems Research Laboratories Inc,簡稱TSRL公司)網站就提供該項服務,查詢網址為:http://www.tsrlinc.com/resources/services/。
4.
溶出儀的校正與校正片使用
現今,各儀器廠商皆已具備對儀器進行機械校正的能力。在這基礎之上,幾乎皆可通過校正片(美G/中G藥典出品)測試。如無法通過,則是溶出杯問題。蓋因在手工燒制底部時,無法均一性地制得內部完整半球形(燒制厚度不一所致),如圖所示。此時,需更換溶出杯后再行測試。故建議使用時,安裝位置、槳桿、溶出杯三者皆應統一固定
[]。
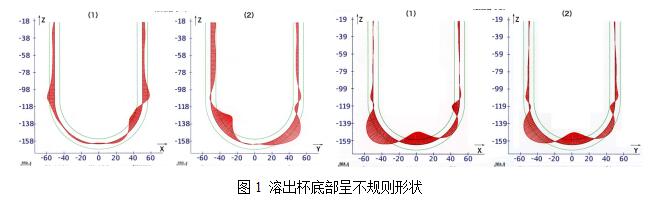
圖1 溶出杯底部呈不規則形狀
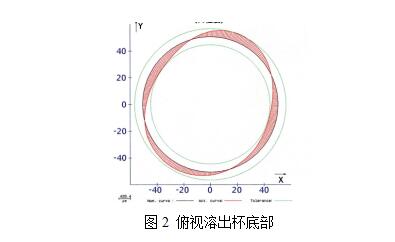
圖2 俯視溶出杯底部
5.
對“漏槽條件”的再認知
漏漕條件的定義為:溶出介質體積應大于溶解藥物主成分(該量為制劑**大規格量)所需體積的**少3倍量,以保證藥物溶出不受其溶解性的顯著影響。該理念是美G學者在設計
溶出儀設備、斟酌溶出杯體積時,根據當時已上市的大部分藥物后設計的折中體積。現今,由于溶出杯體積已固定(900~1000ml),因此若以此為出發點,根據某藥物在某溶出介質中的溶解度來推算采用“何種溶出介質”或“添加多少濃度的表面活性劑”,將極大地忽視“藥物制劑”作用,即是對“藥劑 —— 就是改善藥物水難溶性程度”這一概念的擯棄。故筆者認為,該理念在現今溶出度試驗應用中已無用武之地。歸根結底,還是要取原研制劑,采用溶出曲線循序漸進般予以分析和辨明。
6.
需擬定溶出度檢查的劑型
除通常的口服片劑與膠囊劑外,難溶性藥物的顆粒劑/散劑亦需擬定溶出度檢查項,因為此種劑型同樣存在主成分在體內釋放、吸收過程
[]。對于咀嚼片,服用時雖要求“咀嚼后吞下”,但考慮到可能有部分患者誤當作普通片,故為保證該情況下藥物仍能在體內快速崩散、溶出,發揮療效,美G藥典中幾乎所有咀嚼片皆擬定了溶出度檢查
[],而我G目前尚未制訂,值得借鑒。
7.
對“質量標準中溶出度試驗所發揮作用”的再認識
質量標準中的溶出度試驗,由于僅是一個介質、一個時間點、一個限度的擬定。故即便經過以上嚴謹、周密、科學、全面的驗證予以了確定,但在全面評價藥物內在品質上仍會捉襟見肘、以偏概全,尤考證不同來源同一制劑品質差異性方面,更顯不足。此時。則強烈推薦進行溶出曲線、乃**多條溶出曲線的測定比對工作。現今,G際衛生組織針對不同來源的同一制劑的質量監測與評價已全面引入此法
[];日本對上市仿制藥的質量評價也將溶出度試驗單列(其他項目仍是由地方藥檢所負責),由G立醫藥品食品衛生研究所(即G家藥品檢驗所)藥品部親**持指導“如何采用多條溶出曲線評價口服固體制劑內在品質”工作,并與《藥品品質再評價工程》的實施相輔相成
[]。
我G藥監部門每年均要進行大量的市場抽查監測檢驗,僅按質量標準檢驗往往不能更深層次反映問題存在,故筆者強烈建議引入該套評價體系。自2008年,G家食品藥品監督管理局開展“G家藥品評價性抽驗工作”以來,已將該理念與作法貫徹并實施,取得顯著效果。
8.
期待與展望
制藥行業作為“高科技行業”的核心體現就是制劑,具體細化**“固體制劑的工業藥劑學”,而該點又是與體外溶出度試驗密不可分、相輔相成的。我們只有把握住這一要點,才能“四兩撥千斤”般地“一針見血”地抓住事物本質,進而推動行業發展整合與優勝劣汰。
感謝閱讀此文的讀者,讓我們攜手來夯實G產仿制藥發展基石,來共同培養G產仿制藥成長土壤。為了G產制劑“勵精圖治、殺出重圍”的那**早日到來,讓我們黽勉同行、砥礪同心!再次感謝您的閱讀!
謝沐風. 改善溶出度評價方法,提高固體藥物制劑水平. 中G醫藥工業雜志 2005,7(36),447~451
謝沐風. 溶出度研究系列(一).中G藥品標準, 2005,6(6):42~46.
謝沐風. 溶出度研究系列(二).中G藥品標準,2006,1(7):48~51.
USP33-NF28 <1092 THE DISSOLUTION PROCEDURE: DEVELOPMENT AND VALIDATION>
FIP Guidelines for Dissolution Testing of Solid Oral Products 和 FIP/AAPS Guidelines for Dissolution/In Vitro Release Testing of Novel/Special Dosage Forms*【G際藥學聯合會(International Pharmaceutical Federation/FIP)頒布的“口服固體制劑試驗指導原則”和“新劑型體外溶出度/釋放度試驗指導原則”】 摘自官方網站(http://www.fip.org → Pharmaceutical Sciences and the FIP Special Interest Groups → Dissolution/Drug Release)
謝沐風. 溶出曲線相似性的評價方法 中G醫藥工業雜志 2009,4(40):
謝沐風、張啟明等. G外藥政部門采用溶出曲線評價口服固體制劑內在品質的情況簡介. 中G藥事,2008,3(22):257-261.
謝沐風. 提高質量標準,促進品質提升,帶動行業發展 —— 議如何促進G產藥品的質量 中G醫藥工業雜志,2007,11(38):
**亞敏. 淺談溶出度檢查方法的建立 摘自G家食品藥品監督管理局藥品審評中心網站【www.cde.org.cn → 電子刊物 → 20071130 淺談溶出度檢查方法的建立】
李廣*,張育平等 水楊酸pKa的紫外_可見分光光度法測定. 河南科技學院學報(自然科學版)2005,1(33),75~77
樊志順,李勤朗 用PH分配原理PKA值對臨床用藥問題的討論分析 實用醫技雜志 1996,2(3) 6~8
晁若冰,伍朝員,賀曉英 弱酸弱堿性藥物PKA值的分光光度測定法 華西藥學雜志1990,5(2) 104~106
張啟明、謝沐風等. 采用多條溶出曲線評價口服固體制劑的內在質量 中G醫藥工業雜志,2009,12(40):946-950
青柳伸男. 日本薬局方溶出規格の設定方針について 獨立行政法人-醫薬品醫療機器総合機構(http://www.pmda.go.jp) → 日本薬局方 → 技術情報【摘自日本G厚生省G家藥品審評中心網站 → 日本藥典 → 技術情報 如何制訂溶出度試驗質量標準/青柳伸男撰寫】
用于艾滋病、瘧疾和結核治療的多來源(仿制)制劑(FPPs)預認證文件提交指導原則(補充1) 摘自世界衛生組織網站【http://www.who.int / → Programmes and projects → Prequalification of Medicines Programme → 點“中文” → 點“繼續” → 點“仿制藥 (Generics)” → 點“補充1”】
英G藥典2008年版
. 鄭G鋼 化學藥品普通口服固體制劑溶出度方法驗證易忽視的幾個問題. G家藥品審評中心-電子刊物-化藥藥物評價-化藥質量控制 編號20071106
唐素芳. 化學藥品溶出度方法研究 摘自G家食品藥品監督管理局藥品審評中心網站【www.cde.org.cn → 電子刊物 → 20040420/化學藥品溶出度方法研究】
劉樹春,劉言,范積芬. 栓劑溶出度檢查方法的研究 天津藥學 2003,2(15):13-16
牛麗紅,陳啟蒙. 卡鉑栓劑溶出度實驗方法研究 天津藥學 2003,5(15):4-6
. 《藥典注釋(中G藥典1990年版二部)》 1131頁 化學工業出版社 1993年出版
. M. Morihara, N, Aoyagi(青柳伸男)* Hydrodynamic Flows Around Tablets in Different Pharmacopeial Dissolution Tests. Drug Development and Industrial Pharmacy, 28(6),655-662(2002)
謝沐風 高效、準確、快捷地測定大批量溶出度樣品. 中G醫藥工業雜志,2010,7(41):
青柳伸男. 溶出試験の変動要因と適格性保証 製剤と機械, 290,2-3 (2003) 【溶出度試驗注意事項與質量標準擬定的科學性與適用性】
謝沐風. 溶出度測定中應注意的若干問題. 中G醫藥工業雜志,2006,12(37):859-862
附件5 — 固定劑量復方制劑注冊指導原則 摘自世界衛生組織網站【http://www.who.int / → Programmes and projects → Prequalification of Medicines Programme → 點“中文” → 點“繼續” → 固定劑量聯合用藥(FDCs)】
何仲貴等譯 (美)沃特貝恩德(Waterbeemd,H.van)等著 《藥物生物利用度》 化學工業出版社 2007年第1版
BP2009年版光盤 以“Prolonged-release”搜索品種名稱即可。
青柳伸男. 日本のジェネリック醫薬品は世界で**も厳しい基準により承認されている 月刊ジェネリック, 6,7-9 (2003)【題目:日本仿制藥質量標準被認為是世界上**嚴格的仿制藥標準】
摘自G家食品藥品監督管理局藥品審評中心網站【www.cde.org.cn → 新聞中心 → 工作動態→ 通知公告 → (第2頁)20110128 CDE網站開通“日本藥品體外溶出試驗信息庫”的通知】
金少鴻,寧保明譯 《世界衛生組織藥品標準**委員會第40次技術報告(世界衛生組織技術報告叢書)》附錄7-多來源(仿制)藥品:建立可互換性注冊要求指導原則 中G醫藥科技出版社 2009年第1版。
新醫薬品の規格及び試験方法の設定について 獨立行政法人-醫薬品醫療機器総合機構(http://www.pmda.go.jp) → G際関係業務 → ICH → Quality品質/品質に関するガイドライン【摘自日本G厚生省G家藥品審評中心網站 → G際協調事務(ICH) → 品質保證 《新藥質量標準中各檢測項目與測定方法擬定指導原則》】
G家食品藥品監督管理局標準WS-802(X-588)-2002 拉米夫定片(葛蘭素史克制藥-蘇州有限公司出品)
Biowaiver monographs 【G際藥學聯合會(International Pharmaceutical Federation/FIP)頒布的“可豁免生物等效性試驗藥物清單”】 摘自官方網站(http://www.fip.org → Pharmaceutical Sciences and the FIP Special Interest Groups → BCS and Biowaiver → Biowaiver monographs)
寧保明,張啟明主譯 漢森(Hanson)等著 《溶出度試驗技術(第3版)》第24和45頁 中G醫藥科技出版社,2007年第1版
謝沐風. 關于擬定水難溶性藥物顆粒劑(口服干混懸劑)溶出度檢查的建議 中G醫藥工業雜志,2006,6(37):
關于咀嚼片溶出度技術要求的探討 藥品技術評價文集(第二輯) 中G醫藥科技出版社 2008年出版 第64項
Theo G. Dekker等著, 作為WHO生產資格預認證(PQ)項目一部分的抗逆轉錄病毒產品的持續性監測 摘自世界衛生組織網站【http://www.who.int / → Programmes and projects → Prequalification of Medicines Programme → 點“中文” → 點“繼續” → 藥品的監測(Ongoing monitoring)文檔】
【G外考察】丁麗霞黨委副書記等三人隨G家食品藥品監督管理局稽查局團組赴日交流(2009-10-30) “G家計劃中對仿制藥質量評價的溶出試驗由G立醫藥品食品衛生研究所負責” 摘自中G食品藥品檢定研究院網站(http://www.nicpbp.org.cn) → 工作動態 → G際交流




